苹果公司的研究人员用一个叫SimpleFold的模型证明,预测蛋白质的三维结构,也许并不需要像AlphaFold2那样复杂的“独门秘籍”,一套通用的变换器(Transformer)架构就可能足够了。
一切要从那个“老大难”问题说起
蛋白质的功能,很大程度上由其精确的三维空间结构决定。搞清楚蛋白质是怎么从一条氨基酸链折叠成复杂立体形态的,是几十年来困扰生物学家的核心问题之一。早在1972年,Christian Anfinsen就因证明了蛋白质的氨基酸序列决定其三维结构而获得诺贝尔奖。可怎么从序列解出结构,这条路走了半个世纪。最初,科学家们只能用X射线晶体学这类又慢又贵的实验方法,像拍分子级照片一样,一点点解析结构。
后来计算机科学发展起来,人们开始尝试用计算来预测。为了检验各路方法的成色,1994年起,全球科学家们每两年举办一次“蛋白质结构预测关键评估”(Critical Assessment of protein Structure Prediction, CASP)大赛,这成了蛋白质折叠领域的“华山论剑”。多年来,各路神仙各显神通,但预测精度始终和实验方法有不小的差距。
直到2020年,DeepMind公司的AlphaFold2在CASP14上一战成名,其预测精度达到了与实验方法相媲美的程度,整个领域为之震动。AlphaFold2的成功,被看作是“炼丹术”的极致。它设计了一套极其复杂且精巧的架构,里面塞满了为蛋白质折叠任务量身定做的“独门秘籍”,比如利用多序列比对(Multiple Sequence Alignments, MSAs)寻找进化亲缘关系,构建显式的“对表示”(pair representation)来描述氨基酸残基之间的关系,还有那个计算量巨大的“三角形更新”(triangular updates)模块来回传递空间信息。
这些设计,就像是给模型焊上了一套专门的“外骨骼”,硬编码了研究人员对蛋白质折叠物理过程的理解。一时间,AlphaFold2的架构成了行业标杆,后来者如华盛顿大学的RoseTTAFold等,也大都沿着这条路子走,不断地在这些领域特有的复杂模块上做文章。大家似乎形成了一个共识:搞定蛋白质折叠,就得用这种重型、特化的武器。
换个思路行不行?
生成模型领域大爆发仍然持续。从文本生成图像的DALL-E、Midjourney,到各种3D模型生成,它们的核心思路出奇的一致:用一个相对通用的架构,喂给它海量的数据,让模型自己去学习从输入到输出的映射规律。它们不依赖于开发者硬塞给它的“先验知识”,而是直接从数据中悟道。那能不能也像生成图像一样,给模型一个氨基酸序列(相当于文本提示),让它直接“画”出蛋白质的三维结构?
一些研究者开始了尝试。他们将扩散(diffusion)和流匹配(flow-matching)这类生成模型技术引入蛋白质折叠。但早期探索者们还是有点“路径依赖”,在架构上依然保留了AlphaFold2那些计算昂贵的组件,比如“对表示”和“三角形更新”。这时,苹果公司的研究人员带来了SimpleFold。
SimpleFold的核心思想是,把蛋白质折叠这个事,彻底看成一个条件生成任务,就像“文本生成图像”一样。它的目标,是从一团随机的高斯噪声出发,在氨基酸序列的指引下,一步步把这团噪声“雕刻”成一个精确的全原子蛋白质结构。这个“雕刻”的过程,就是由一种叫做“流匹配”(flow-matching)的技术来控制的。
你可以把流匹配想象成一个时间依赖的变换过程。在时间t=0时,我们有一堆杂乱无章的原子坐标(高斯噪声)。在时间t=1时,我们希望得到一个真实、有序的蛋白质结构。流匹配模型要学习的就是一个速度场,它告诉每个原子在从0到1的每一个瞬间,应该朝哪个方向移动,移动多快,最终才能到达正确的位置。
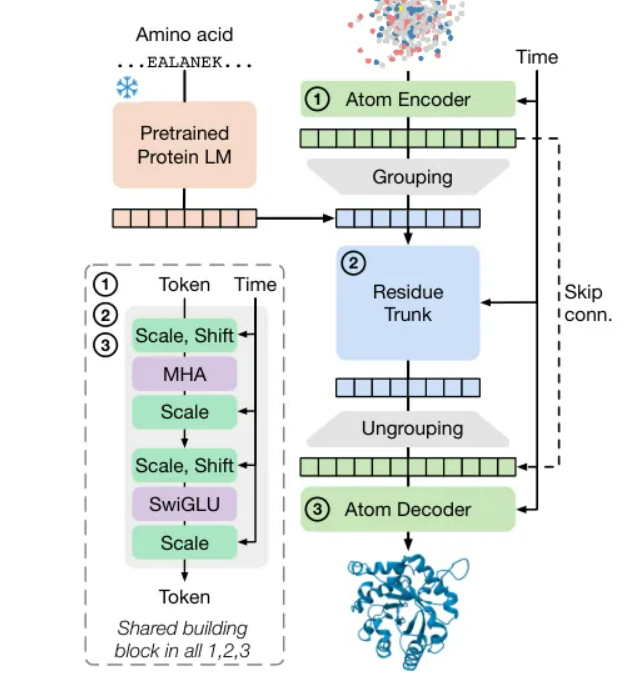
上图就是它的架构图,极其清爽。没有MSA,没有“对表示”,没有“三角形更新”,什么都没有。它只用了最基础、最通用的变换器模块。
整个架构是一个“细-粗-细”的流程:
原子编码器(细):输入的是带噪声的原子坐标和原子类型等特征。这个编码器只关注局部,也就是一个原子只跟它周围的邻居们互动。处理完后,它会把同一个氨基酸残基里的所有原子信息“打包”(平均池化),生成一个代表这个残基的“残基标记”。
残基主干(粗):这是模型的主体和计算核心。它接收上一步打包好的“残基标记”,同时,它还接收一个来自预训练蛋白质语言模型ESM2-3B的序列嵌入。ESM2就像一个“蛋白质专家”,它已经读过了海量的蛋白质序列,能深刻理解序列中蕴含的生物学信息。这两部分信息合并后,在这个巨大的变换器主干网络里进行充分的信息交互和推理。
原子解码器(细):残基主干处理完毕后,输出带有结构信息的残基标记。这些标记被“解包”,重新投射回每个原子身上。原子解码器再对这些原子信息进行一轮局部的精细调整,最终输出预测的速度场,也就是指导原子如何移动的指令。
就是这么简单。它把处理旋转对称性的难题,也用一种最“暴力”的方式解决了:在训练时,把目标蛋白质结构随机转来转去,让模型自己去学习这种旋转不变性,而不是设计复杂的等变网络。为了让这套简单的架构发挥最大威力,SimpleFold走了另一条路:用海量数据来喂养。
研究人员混合了三类数据:首先是PDB(蛋白质数据库)里经过实验验证的约16万个高质量结构;其次是从AlphaFold数据库的SwissProt子集中筛选出的约27万个高质量预测结构;最后,也是最大的一头,是来自AFESM数据库的,经过筛选和去重的近200万个代表性结构。而对于最大的30亿参数模型,他们更是将数据集扩充到了惊人的近900万个结构。
SimpleFold的实际表现
研究人员在两个公认的蛋白质折叠基准测试CAMEO22和CASP14上,对SimpleFold进行了一系列评估。
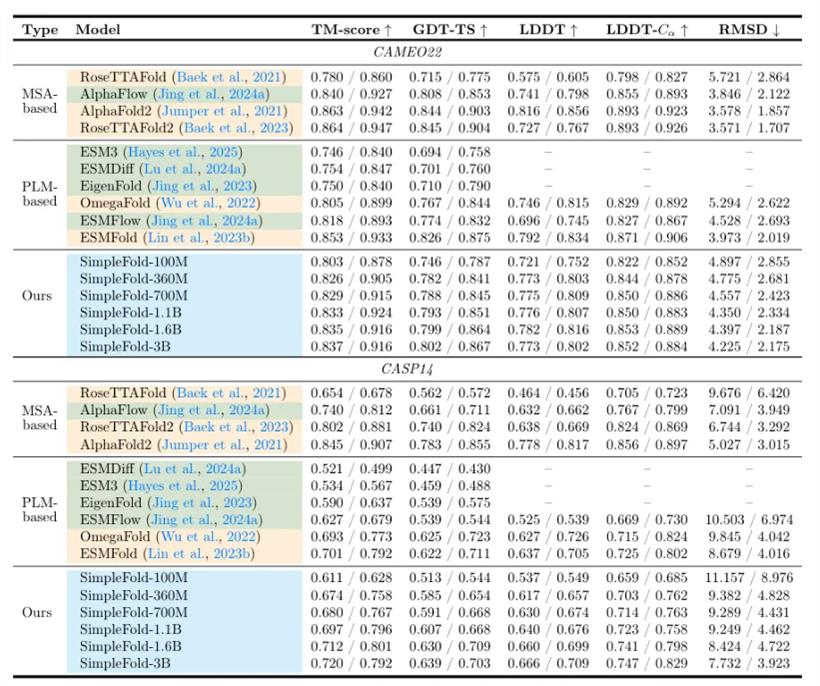
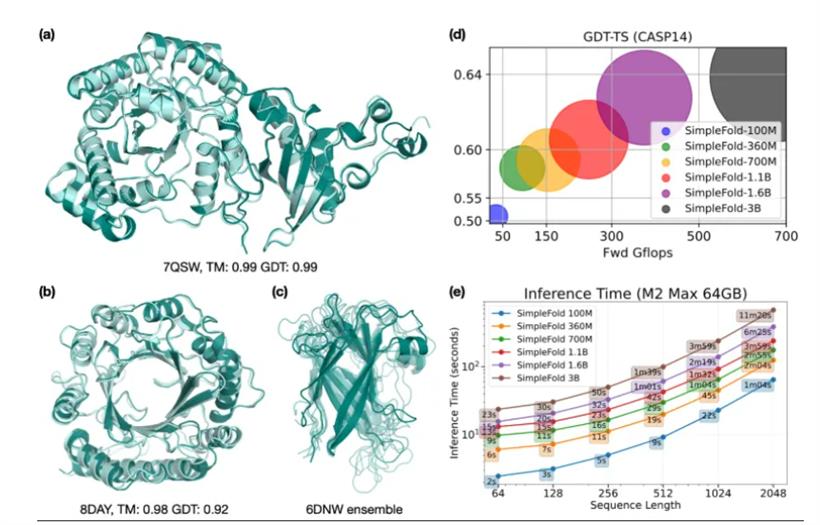
从上表可以看出,尽管架构简单,但SimpleFold-3B的表现极具竞争力。在CAMEO22上,它的各项指标几乎与ESMFold和RoseTTAFold2持平,达到了AlphaFold2性能的95%以上。在更具挑战性的CASP14上,它甚至超过了同样使用PLM(蛋白质语言模型)的ESMFold。这证明,放弃那些复杂的领域专用模块,并不会导致性能断崖式下跌。一条更简洁、更通用的路径是完全可行的。
SimpleFold真正的杀手锏,还在于它的“生成”能力。传统的回归模型,比如AlphaFold2,对一个序列只会给出一个确定性的预测结构。但生物世界里,蛋白质不是僵硬的,它会运动、会变化,甚至有些蛋白质本身就存在多种稳定构象。
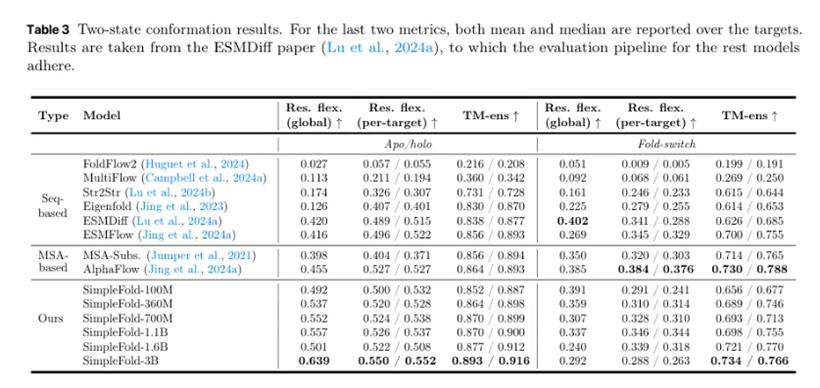
在预测具有多种构象的蛋白质(Apo/holo和Fold-switch数据集)时,SimpleFold同样表现出色,甚至在Apo/holo上取得了当前最佳的性能。这对于理解蛋白质功能和药物发现至关重要。
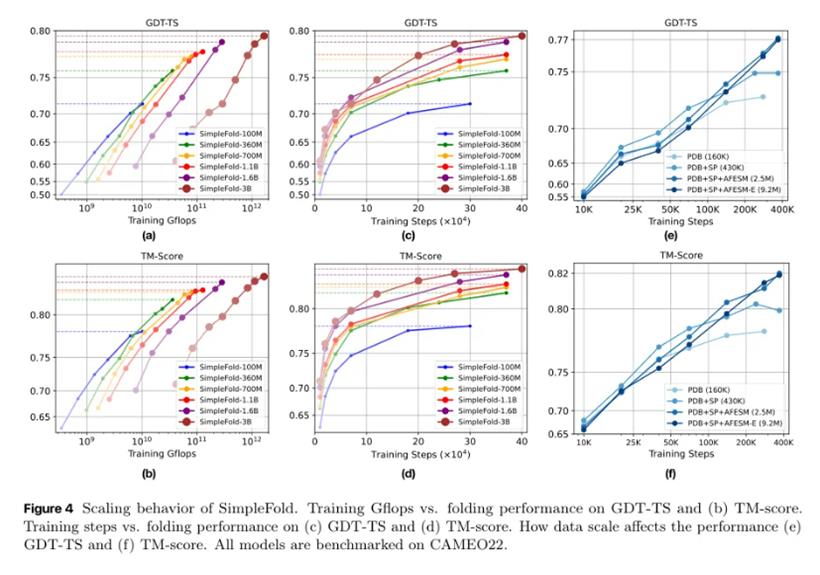
SimpleFold完美展现了“规模效应”(Scaling Law)的魅力。无论是增加模型参数量(从1亿到30亿),还是扩大训练数据集(从16万到近900万),模型的性能都随之稳步提升。这说明SimpleFold的通用架构设计是一条康庄大道,未来的提升路径清晰明了:更大的模型,更多的数据。通过拥抱生成模型的简洁哲学,并借助海量数据和计算的力量,我们同样可以抵达甚至超越过去的高度。
参考资料:
https://github.com/apple/ml-simplefold
https://arxiv.org/abs/2509.18480


 闽公网安备 35020302035485号
闽公网安备 35020302035485号






